EdgeOne Medical - Blog
Our platform for fostering conversation on product development and product lifecycle management topics of interest with the community.
published on February 28, 2015 by
FDA’s final rule on combination products (21 C.F.R. Part 4) set forth current good manufacturing practice requirements that must be met for the entire product (drug/device or biologic/device), regardless if the device design occurs internally or externally to the Marketing Authorization Holder (MAH). Although many elements of CP cGMPs are well understood, it appears many are focused on understanding design control and supplier evaluation in the context of the new regulation and guidance.

In our experience, small to medium size pharma-centric, CP companies that do not maintain device development expertise in-house must now identify the appropriate partner to first develop the device and second, create a compliant design history file. This scenario above is troubling because these two facets of the cGMPs are not well understood in small and medium size pharma-centric CP companies.
Let’s take a fresh look at how the precedent has been set--- the FDA issued a Warning Letter to Amgen in early 2013 citing deficiencies in their Design Control process. This Warning Letter highlights FDA’s willingness to enforce combination products, regardless if they are market leaders in the space. So is this case a roadmap for things to come? Based on FDA’s recent Guidance and industry discussions on this topic, it is highly likely your firm will be tasked with defending your CP DHF and supplier qualification activities.
The following are key points the MAH should consider when outsourcing device design and development activities.
- MAH should ensure that they implement their own internal processes that interface with and account for outsourced party’s implemented processes (820.30, MDD, 13485, and 14971).
- Design History Files and Risk Management files should link to the monitoring of post-market activities as seen in Figures A and B.
- Robust design and proper documentation created throughout the development cycle, post market activities will have an adverse effect on product branding and availability.
- CP designed utilizing design control and supported by a DHF builds needed credibility with regulators.
Hence, it is critical to ensure third party designers understand the full design landscape from concept to launch, to post-market, in order to reduce post market “actions” (complaints, Medical Device Reports, Adverse Events, CAPAs, recalls, redesigns, and re-submissions), which will lead to improved audit outcomes.
Figure A: Design and Risk File Foundation and its Post-Market Launch Quality System Dependencies
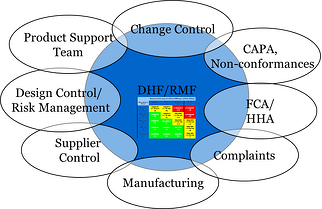
The License holder maintains ultimate responsibility for licensure, and thus, in making decisions regarding product design changes. A continual flow of information to and from the MAH is critical to ensure product is safe and efficacious for its intended use post-launch (see Figure B below). Based on post-market data monitoring, the MAH will have responsibility to decide when product enhancements should occur. Hence, MAH engagement with qualified suppliers will be imperative in the post-market phase. Figure B below depicts the post market activities between the MAH and their external suppliers.
Figure B- Activities and Engagement During Post-Launch (Example)
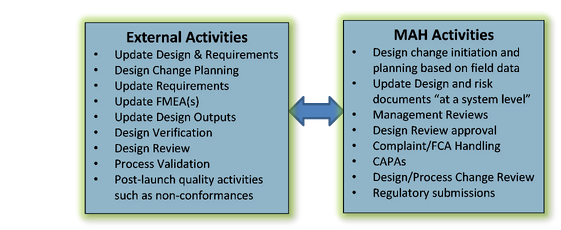
Conclusion: Compliance with FDA’s CP regulation and cGMP Guidance documents must consider the product design documentation generated by an external provider as an extension of their design and development process. If you don’t have the expertise in this area, enlist the support of a third party expert. FDA’s audit expectation is that the MAH has control over the product’s design regardless if conducted internally or externally. Partnering with device firms that understand the risks to a MAH will inevitably create a relationship predicated on mutual understanding of compliance to the regulations in which both companies share development and post-market responsibilities when FDA arrives.
EdgeOne Medical and the leaders of their Combination Product Development Services Team have been supporting the device development component of Drug/Biologic and Device Combination Product Program for the past fifteen years. Services include Strategic Product Development, Project Management, Regulatory Strategy & Submission, Quality Systems, Remediation, Risk Management, and Device Product Testing.
